01 October 2021: Original Paper
Outcomes After Living Donor Liver Transplantation in Pediatric Patients with Inherited Metabolic Diseases
Yukihiro Sanada1ABCDEF*, Yasunaru Sakuma1DF, Yasuharu Onishi1B, Noriki Okada1B, Naoya Yamada1B, Yuta Hirata1B, Go Miyahara1B, Takumi Katano1B, Toshio Horiuchi1B, Takahiko Omameuda1B, Alan Kawarai LeforDOI: 10.12659/AOT.932994
Ann Transplant 2021; 26:e932994






Abstract
BACKGROUND: There is no consensus about the long-term prognosis of pediatric patients with a variety of rare liver diseases but with inherited metabolic diseases (IMDs). We retrospectively reviewed the developmental outcomes of patients with IMDs undergoing living donor liver transplantation (LDLT).
MATERIAL AND METHODS: Between May 2001 and December 2020, of 314 pediatric patients who underwent LDLT, 44 (14%) had IMDs. The median age at LDLT was 3.0 years old (range 0-15.0 years). Associations between the post-transplant complications and graft survival rate in patients with IMDs and biliary atresia (BA) were calculated. We evaluated the safety of LDLT from heterozygous carrier donors, the prognosis of patients with IMDs who have metabolic defects expressed in other organs, and developmental outcomes of patients with IMDs.
RESULTS: The 10-year graft survival rates in patients with IMDs and BA were 87% and 94%, respectively (P=0.041), and the causes of graft failure included pneumocystis pneumonia, acute lung failure, hemophagocytic syndrome, hepatic vein thrombosis, portal vein thrombosis, and sepsis. The rate of post-transplant cytomegalovirus viremia in patients with IMDs was higher than that of patients with BA (P=0.039). Of 39 patients with IMDs, 15 patients (38%) had severe motor and intellectual disabilities in 4 patients, intellectual developmental disorders including epilepsy in 2, and attention-deficit hyperactivity disorder in 2. Of 28 patients with IMDs, 13 (46%) needed special education.
CONCLUSIONS: The long-term outcomes of LDLT in patients with IMDs are good. However, further long-term social and educational follow-up regarding intellectual developmental disorders is needed.
Keywords: Brain Diseases, Metabolic, Inborn, Liver Cirrhosis, Biliary, Liver Transplantation, Adolescent, Child, Child, Preschool, Female, Graft Survival, Humans, Infant, Infant, Newborn, Living Donors, Metabolic Diseases, Postoperative Complications
Background
Liver transplantation (LT) is an established curative treatment for pediatric patients with end-stage liver diseases such as liver cirrhosis. The reported incidence of LT for patients with inherited metabolic diseases (IMDs) is 8.0–20.2% [1–9]. It has been reported that the 5-year and 10-year graft survival rates in patients with IMDs were 73.1–94.5% [1–5,7–9] and 62.0–90.0% [2–4,7,8], respectively. However, there are many problems surrounding LT for patients with IMDs. Although there is a possibility of heterozygous carrier donors in the case of living donor liver transplantation (LDLT) for patients with IMDs, there is no consensus about the safety of LDLT from heterozygous carrier donors [10–13]. Although the prognosis of patients with urea cycle diseases is good because LT is curative, the prognosis of patients with IMDs who have metabolic defects expressed in other organs remains undefined [3,14–18]. In addition, although there are potential intellectual developmental disorders and neurological sequelae in patients with IMDs and hyperammonemia, the outcomes for these patients are unclear.
We present a retrospective analysis of our experience performing LDLT on patients with IMDs, focusing on their long-term prognosis and associated intellectual developmental problems.
Material and Methods
PATIENTS:
Between May 2001 and December 2020, 314 LDLTs were performed on pediatric patients with end-stage liver disease, acute liver failure, and IMDs in our institution. Of these, 221 patients with biliary atresia (BA) and 44 patients with IMDs underwent LDLT; these patients were included in this study. Forty-four patients (14%) had IMDs including ornithine transcarbamylase deficiency (OTCD) (n=19), Wilson’s disease (n=5), neonatal hemochromatosis (n=5), maple syrup urine disease (n=4), methylmalonic acidemia (MMA) (n=3), progressive familial intrahepatic cholestasis type 1 (n=2), and citrullinemia (n=2), as well as cystic fibrosis, carbamoyl phosphate synthetase 1 deficiency, Niemann-Pick disease type C, and glycogen storage disease type Ia (each n=1). The indications for LT in patients with IMDs were improvement of quality of life of patients with severe disease manifestations or life-threatening metabolic decompensations despite medical and dietary management. The mean observation time between May 2001 and December 2020 was 10.6±5.4 years. Demographic data for recipients and graft information are shown in Table 1. Approval to conduct this study was obtained from the university clinical research ethics review board at our university (CU No. 20-001).
SURGICAL PROCEDURE OF LDLT:
The type of donor hepatectomy was determined based on the recipient’s standard liver volume, recipient’s weight, and graft volume by preoperative computed tomographic volumetry [19,20]. The donor’s biliary anatomy was evaluated performing intraoperative real-time cholangiography 3 times to define the biliary anatomy, determine the biliary transection line, and identify biliary leakage. A routine donor hepatectomy was performed using intraoperative ultrasonic guidance. The donor’s left hilar plate was transected using a scalpel.
For the recipient operation, inverted T-shape incisions were used, and total hepatectomy was performed. In many infants, after total hepatectomy, the recipient’s right, middle, and left hepatic veins became a single orifice, and the recipient’s hepatic vein was anastomosed to the graft’s hepatic vein. The recipient’s portal vein was anastomosed to the graft’s left portal vein. Hepatic artery reconstruction was performed using microsurgical techniques. Biliary reconstruction was performed using a Roux-en-Y hepaticojejunostomy.
IMMUNOSUPPRESSION THERAPY:
Tacrolimus and methylprednisolone were used as standard post-operative immunosuppression therapy. The target trough level of tacrolimus was gradually decreased. Mycophenolate mofetil was used when more potent immunosuppression was required; for example, in ABO-incompatible recipients, in patients with acute cellular rejection episodes, or in patients with liver dysfunction after the cessation of methylprednisolone therapy.
POST-TRANSPLANT MANAGEMENT:
During the post-transplant period, patients routinely received anticoagulants and underwent Doppler ultrasonography. Anticoagulation treatment was started with intravenous dalteparin sodium (100 U/kg/day) several days postoperatively. If hepatic inflow and outflow were sufficient, we usually withdrew anticoagulant at post-operative day (POD) 14. Doppler ultrasonography was used for follow-up imaging surveillance. Doppler ultrasonography was performed routinely twice per day until hospital discharge, and thereafter at 1, 3, 5, and 9 months, and then every 6 months after LDLT.
In our department, surveillance for infection is based on peripheral blood studies. Serum cytomegalovirus antigenemia (C7-HRP) and β-D-glucan were performed routinely once per week until hospital discharge, and monthly thereafter following LDLT. Prophylactic treatments due to antiviral and antifungal agents were performed if the serum C7-HRP and β-D-glucan was positive.
STATISTICAL ANALYSIS:
The significance of differences between 2 groups was evaluated using the chi-squared test and Mann-Whitney U test. Associations between the recipient, graft, and post-transplant complications were evaluated using univariate analysis. Graft survival rates were calculated by the Kaplan-Meier product-limited method, and differences in survival between the 2 groups were then compared using the log-rank test. All statistical analyses were performed using EZR (Saitama Medical Center, Jichi Medical University, Saitama, Japan), which is a graphical user interface for R (The R Foundation for Statistical Computing, Vienna, Austria), and differences were considered to be significant with values of
Results
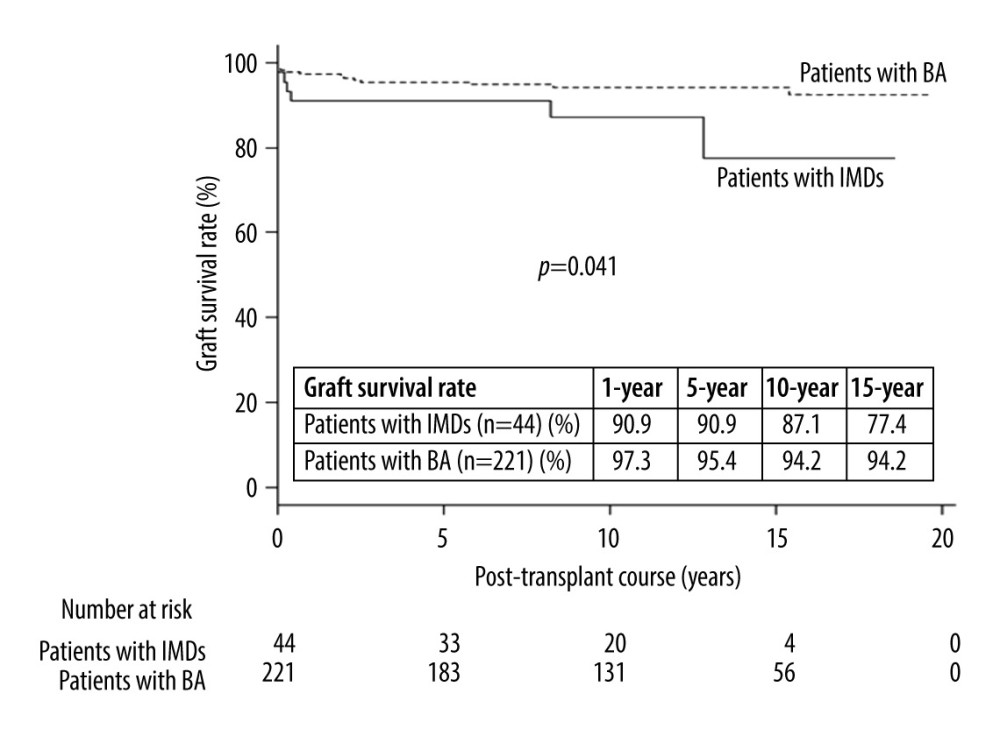
GRAFT SURVIVAL RATES:
The 10-year graft survival rates in patients with IMDs and BA were 87% and 94%, respectively (P=0.041) (Figure 1). The causes of graft failure included pneumocystis pneumonia, acute lung failure, hemophagocytic syndrome, hepatic vein thrombosis, portal vein thrombosis, and sepsis in patients with IMDs. Of the patients with graft failure, 2 patients underwent repeat LT. One patient with graft failure due to hepatic vein thrombosis underwent deceased donor liver transplantation 8.2 years after the first LDLT and did well. Another patient with graft failure due to portal vein thrombosis underwent repeat LDLT on POD 15 and died of sepsis. The causes of graft failure included chronic rejection in 5 patients with BA, bowel perforation in 2, bowel perforation in 2, acute encephalitis in 2, cerebral hemorrhage in 1, hepatic vein thrombosis in 1, veno-occlusive disease in 1, and sepsis in 1. Of the 7 patients with graft failure due to chronic rejection, hepatic vein thrombosis, and veno-occlusive disease, all patients underwent repeat LT.
ANALYSIS OF RISK FACTORS FOR POST-TRANSPLANT COMPLICATIONS IN PATIENTS WITH IMDS AND BA:
The rate of post-transplant acute cellular rejection in patients with IMDs was lower than that with BA (27% vs 44%, P=0.044) (Table 2). The rate of post-transplant cytomegalovirus viremia in patients with IMDs was higher than that with BA (50% vs 33%, P=0.039) (Table 2). Serological patterns of cytomegalovirus infection in patients with IMDs (n=43) were Donor (D)+/Recipient (R)+ in 17 patients, D+/R− in 12, D−R+ in 6, and D−R− in 8. Serological patterns of cytomegalovirus infection in patients with BA (n=219) were D+/R+ in 111 patients, D+/R− in 51, D−R+ in 20, and D−R− in 37.
LDLT FOR PATIENTS WITH OTCD USING A GRAFT FROM A HETEROZYGOUS CARRIER DONOR:
Relationships between donor and recipient in patients with OTCD (n=19) were father in 12 patients, mother without heterozygous carrier in 5, mother with heterozygous carrier in 1, and maternal aunt without heterozygous carrier in 1. One male patient with OTCD had indications for LT because his disease was the neonatal-onset type (day8, NH3 901 μmol/L) and was resistant to medical treatment at 3 years of age (Table 3, OTCD Case 5) [13]. Except for the OTCD carrier mother, there were no voluntary donor candidates. His mother was doing well and never had any symptoms suggesting hyperammonemia. In addition, her serum ammonia level was normal, and the OTC activity in her liver was 104.4%. He underwent LDLT using a left lateral segment graft from a heterozygous carrier donor at 3.4 years old. The graft volume was 244 g. The donor’s remnant liver volume 81.8%, and graft volume/standard liver volume ratio (GV/SLV) was 54.0%. The post-transplant clinical course was uneventful and they are both doing well without intellectual developmental disorders at 11.4 years after LDLT.
LONG-TERM OUTCOMES OF LDLT IN PATIENTS WITH IMDS WHO HAVE METABOLIC DEFECTS EXPRESSED IN OTHER ORGANS:
Our experience with LDLT for patients with methylmalonic acidemia is shown in Table 4. Three patients with methylmalonic acidemia have normal renal function with restricted diets, but the long-term renal prognosis is not clear. Two patients have growth and developmental disorders, but 1 patient who underwent LDLT at 2 months old has no growth or developmental disorders. One female patient with acute liver failure underwent LDLT using a segment 2 mono-segment graft from her father at 59 days of age. She had progressive neurologic dysfunction and required long-term ventilatory support with a tracheostomy and nutrition via gastrostomy. The period of diagnosis of Niemann-Pick disease type C was 1.6 years after LDLT. She has progressive neurologic dysfunction, Niemann-Pick disease type C cell infiltration of the graft liver, and secondary Crohn’s disease at present [21]. One patient with cystic fibrosis died of progressive respiratory failure 12 years after LDLT.
OUTCOMES OF YOUNGER SIBLINGS AFTER LDLT IN PATIENTS WITH IMDS:
Eleven patients with IMDs who had younger siblings after LDLT included patients with neonatal hemochromatosis (n=4), OTCD (n=3), progressive familial intrahepatic cholestasis type 1 (n=1), carbamoyl phosphate synthetase 1 deficiency (n=1), MMA (n=1), and Niemann-Pick disease type C (n=1). Of the younger siblings of 4 patients with neonatal hemochromatosis, 2 underwent antenatal maternal high-dose immunoglobulin treatment to prevent neonatal hemochromatosis [22]. However, 1 child without immunoglobulin treatment who underwent LDLT from the puerperal mother developed neonatal hemochromatosis and another did not develop neonatal hemochromatosis. In younger siblings of patients with OTCD, 1 carrier received medical treatment and 1 neonate with OTCD underwent DDLT. In younger siblings of patients with carbamoyl phosphate synthetase 1 deficiency, MMA, and Niemann-Pick disease type C, 3 had prenatal amniotic fluid analysis and were healthy.
CURRENT STATE OF INTELLECTUAL DEVELOPMENTAL DISORDERS IN PATIENTS WITH IMDS:
Of 39 patients with IMDs (excluding 5 patients who have died), 15 patients (38%) had severe motor and intellectual disabilities in 4 patients, intellectual developmental disorders including epilepsy in 2, and attention-deficit hyperactivity disorder in 2. Of 28 patients with IMDs (excluding 6 workers and 5 preschool patients), 13 (46%) needed special education.
Discussion
The reported rate of LT in patients with IMDs is 8.0–20.2% [1–9]. The 5-year and 10-year graft survival rates in patients with IMDs are reportedly 73.1–94.5% [1–5,7–9] and 62.0–90.0% [2–4,7–8], respectively (Table 5). Few patients with BA undergo LT and there is a wide variation of outcomes among institutions. In this study, the 10-year graft survival rates in patients with IMDs and BA were 87% and 94%, respectively (
There are many problems associated with LT for patients with IMDs. The indications for LT in patients with IMDs who have metabolic defects expressed in other organs are to preserve life, prevent life-threatening complications, and improve the quality of life (release from restricted diets and prevention of developmental disorders). However, LT for patients with IMDs who have metabolic defects expressed in other organs is not a curative treatment [3,14–18]. Therefore, adequate informed consent regarding the benefits and risks of LT, including the possibility of poor outcomes after LT, should be obtained in each case. In patients with MMA, continuing metabolic damage to the kidneys and brain may occur even after successful LT [23,24]. It has been reported that LT should be performed as a therapeutic option in the early stages of the disease because LT allows prevention of decompensation episodes, normalization of dietary protein intake, and a marked improvement in quality of life [25]. In this study, the 1 patient who underwent LDLT at 2 months had no growth or developmental disorders (Table 4). In patients with Niemann-Pick disease type C, neonatal-onset Niemann-Pick disease type C often presents with jaundice and hepatosplenomegaly from birth, and rarely progresses to liver failure. Therefore, patients with neonatal-onset Niemann-Pick disease type C may require simultaneous diagnosis and treatment [21]. Sufficient informed consent about the chance of post-transplant diagnosis of Niemann-Pick disease type C and poor neurological prognosis should be obtained before LT. We diagnosed Niemann-Pick disease type C 1.6 years after LDLT, and she had progressive neurologic dysfunction requiring long-term ventilatory support with a tracheostomy and nutrition via gastrostomy. She has progressive neurologic dysfunction, Niemann-Pick disease type C cell infiltration of the graft liver, and secondary Crohn’s disease at present [21]. Cystic fibrosis is a multisystem disease caused by mutations in the cystic fibrosis transmembrane conductance regulator gene. Mutations of this gene manifest as epithelial cell dysfunction in the airways, biliary tract, pancreas, gut, sweat glands, paranasal sinuses, and genitourinary tract. Viscous, inspissated bile causes ductal obstruction and hepatotoxicity from retained bile components, leading to fibrosis and ultimately cirrhosis, known as cystic fibrosis liver disease. LT is indicated in patients with decompensated liver cirrhosis. However, pulmonary disease is the main cause of morbidity and mortality in patients with cystic fibrosis [26]. One patient with cystic fibrosis in this series died of progressive respiratory failure 12 years after LDLT. Therefore, before undergoing LT, families need to be informed of the possibility of poor pulmonary outcomes.
Although heterozygous carrier donors are possible when performing LDLT for patients with OTCD, there is no consensus about the safety of LDLT from heterozygous carrier donors (Table 3) [10–13]. Rahayatri et al reported that the recipients have hyperammonemia after LDLT using grafts from asymptomatic heterozygous carrier donors [12]. Therefore, they concluded that the use of grafts from asymptomatic OTCD heterozygous donors in LDLT is acceptable with careful evaluation, and an OTCD heterozygous carrier donor should be avoided if there is another donor candidate. However, poor outcomes of DDLT from donors with unrecognized OTCD have been reported [27–29]. In our heterozygous carrier donor of OTCD, graft OTC activity of liver and GV/SLV was 104.4% and 54.0%, respectively [13]. Sufficient graft OTC activity of the liver and GV/SLV may be required. In addition, sufficient remnant volume for OTCD heterozygous carrier donors is required. We suggest that a donation from an OTCD heterozygous carrier donor should be avoided if there are another living donor candidate or deceased donor.
Information about disease incidence should be given to parents, but there are no specific recommendations. If a woman has an unplanned pregnancy without having received sufficient information, she may choose to undergo an abortion. In this study, of the younger siblings of 4 patients with neonatal hemochromatosis, 2 were born after the mother received antenatal maternal high-dose immunoglobulin treatment to prevent neonatal hemochromatosis [22]. One child whose mother did not receive immunoglobulin treatment who underwent LDLT from the puerperal mother developed neonatal hemochromatosis. Antenatal maternal high-dose intravenous immunoglobulin treatment has been reported to be effective for preventing neonatal hemochromatosis recurrence [30]. In younger siblings of patients with OTCD, carbamoyl phosphate synthetase 1 deficiency, MMA, and Niemann-Pick disease type C, 4 of the mothers had prenatal amniocentesis. Three siblings were healthy, and 1 sibling had OTCD and underwent DDLT. Therefore, information about antenatal maternal high-dose intravenous immunoglobulin treatment and prenatal amniocentesis should be carefully provided to the mother before pregnancy. In addition, genetic counseling will be required for the parents and recipients in the future. However, if siblings undergo LT for IMDs, it has been reported that later siblings should be listed and transplanted at a significantly younger age [31].
There is a potential for intellectual developmental disorders and neurological sequelae in patients with IMDs and hyperammonemia. However, the precise risk is unclear. In the present study, of 39 patients with IMDs (excluding 5 patients who have died), 15 patients (38%) had severe motor and intellectual disabilities in 4 patients, intellectual developmental disorders including epilepsy in 2, and attention-deficit hyperactivity disorder in 2. In addition, of 28 patients with IMDs, 13 (46%) need special education. Kido has been reported that LT had limited effect for ameliorating neurodevelopmental outcome in patients with severe urea cycle diseases because hyperammonemia at the onset time already had a significant impact on the brain [32,33]. Therefore, novel neuroprotective measures should be developed to achieve better neurodevelopmental outcomes in these patients [32]. Moreover, Kido suggested that LT should be considered in patients with urea cycle diseases with maximum ammonia concentration <300 μmol/L at the time of disease onset to protect the brain [34]. In addition, further long-term social and educational follow-up for intellectual developmental disorder is needed in patients with IMDs after LT.
Conclusions
The long-term outcomes of LDLT in patients with IMDs are good. However, the long-term outcomes of LDLT in patients with IMDs who have metabolic defects expressed in other organs remain undefined and further follow-up is needed. In addition, long-term social and educational follow-up for intellectual developmental disorder is needed. This was a retrospective analysis from a single center. Continued evaluation of outcomes and accumulation of further experience are necessary.
Tables
Table 1. Demographic data for recipients and graft information.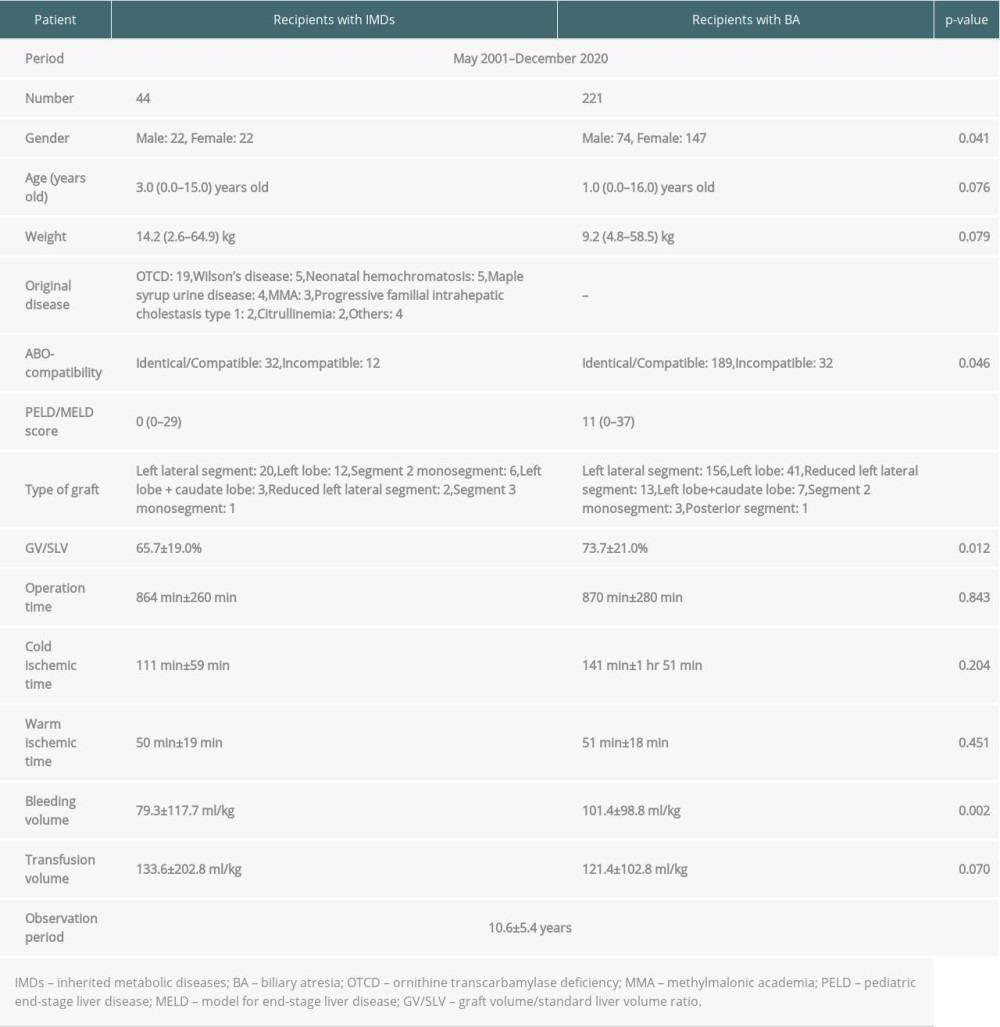 Table 2. Univariate analysis of risk factors for post-transplant complications in recipients with IMDs and BA.
Table 2. Univariate analysis of risk factors for post-transplant complications in recipients with IMDs and BA.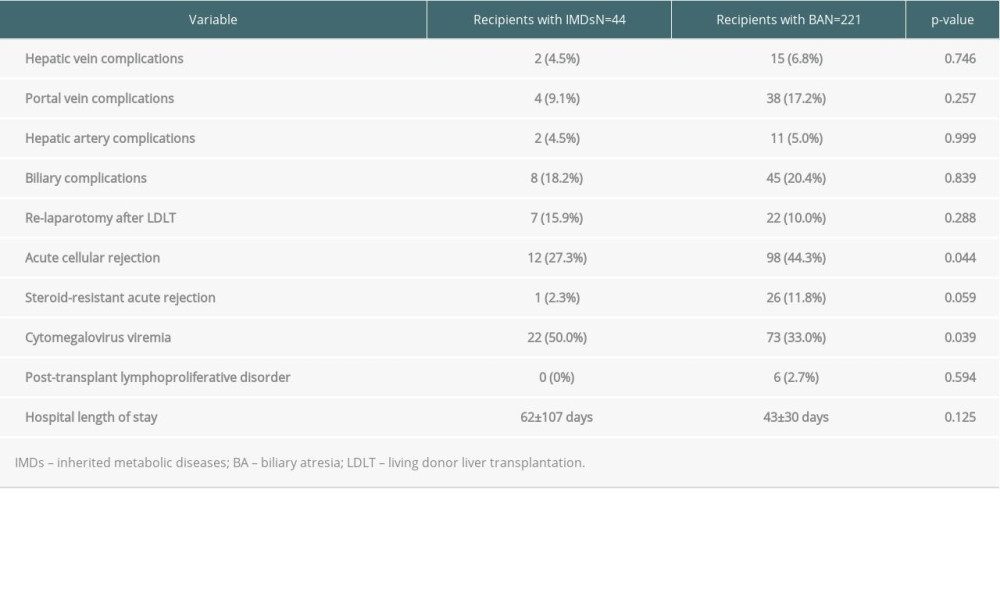 Table 3. Outcomes of LDLT using grafts from OTCD heterozygous carrier donors.
Table 3. Outcomes of LDLT using grafts from OTCD heterozygous carrier donors.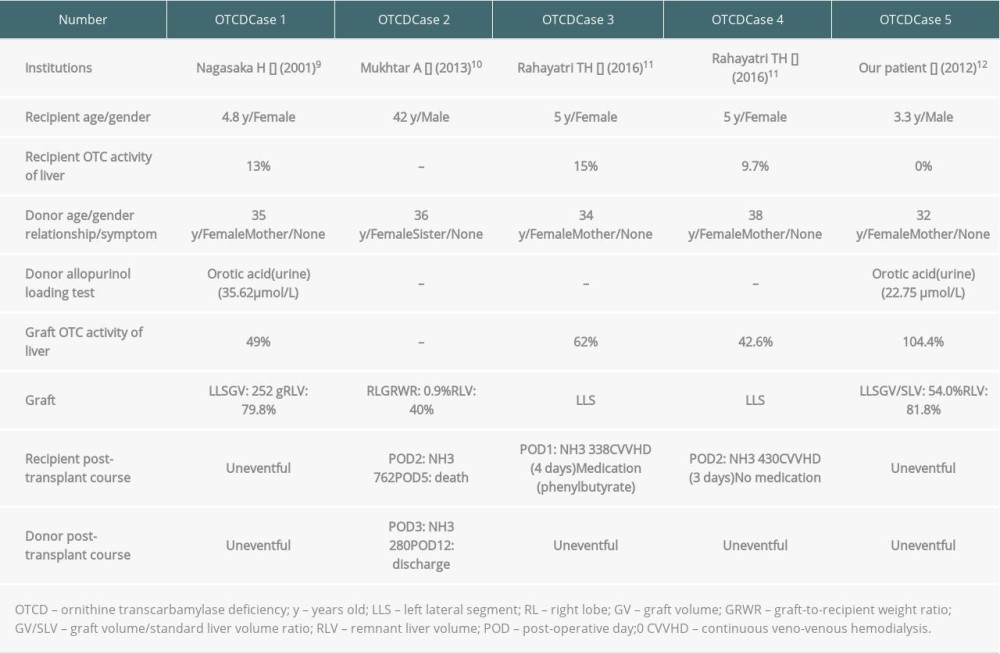 Table 4. Outcomes of LDLT for patients with MMA in our institution.
Table 4. Outcomes of LDLT for patients with MMA in our institution.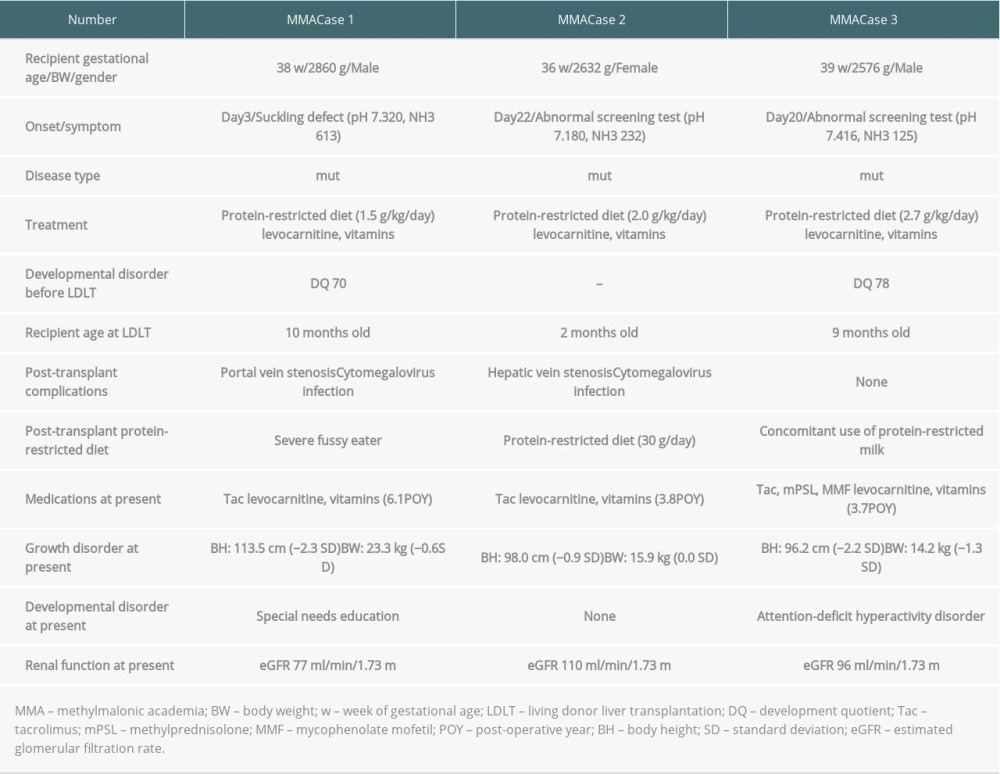 Table 5. Post-transplant survival rates of patients with IMDs.
Table 5. Post-transplant survival rates of patients with IMDs.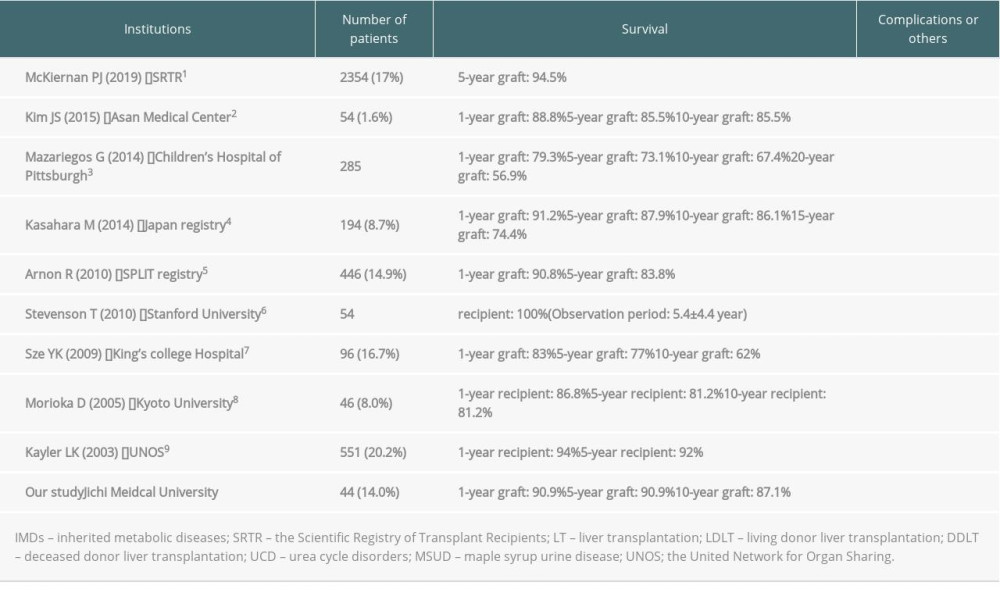
References
1. McKiernan PJ, Ganoza A, Squires JE, Evolving trends in liver transplant for metabolic liver disease in the United States: Liver Transpl, 2019; 25(6); 911-21
2. Kim JS, Kim KM, Oh SH, Liver transplantation for metabolic liver disease: experience at a living donor dominant liver transplantation center: Pediatr Gastroenterol Hepatol Nutr, 2015; 18(1); 48-54
3. Mazariegos G, Shneider B, Burton B, Liver transplantation for pediatric metabolic disease: Mol Genet Metab, 2014; 111(4); 418-27
4. Kasahara M, Sakamoto S, Horikawa R, Living donor liver transplantation for pediatric patients with metabolic disorders: The Japanese multicenter registry: Pediatr Transplant, 2014; 18(1); 6-15
5. Arnon R, Kerkar N, Davis MKSPLIT Research Group, Liver transplantation in children with metabolic diseases: The studies of pediatric liver transplantation experience: Pediatr Transplant, 2010; 14(6); 796-805
6. Stevenson T, Millan MT, Wayman K, Long-term outcome following pediatric liver transplantation for metabolic disorders: Pediatr Transplant, 2010; 14(2); 268-75
7. Sze YK, Dhawan A, Taylor RM, Pediatric liver transplantation for metabolic liver disease: Experience at King’s College Hospital: Transplantation, 2009; 87(1); 87-93
8. Morioka D, Kasahara M, Takada Y, Living donor liver transplantation for pediatric patients with inheritable metabolic disorders: Am J Transplant, 2005; 5(11); 2754-63
9. Kayler LK, Rasmussen CS, Dykstra DM, Liver transplantation in children with metabolic disorders in the United States: Am J Transplant, 2003; 3(3); 334-39
10. Nagasaka H, Yorifuji T, Egawa H, Successful living-donor liver transplantation from an asymptomatic carrier mother in ornithine transcarbamylase deficiency: J Pediatr, 2001; 138(3); 432-34
11. Mukhtar A, Dabbous H, El Sayed R, A novel mutation of the ornithine transcarbamylase gene leading to fatal hyperammonemia in a liver transplant recipient: Am J Transplant, 2013; 13(4); 1084-87
12. Rahayatri TH, Uchida H, Sasaki K, Hyperammonemia in ornithine transcarbamylase-deficient recipients following living donor liver transplantation from heterozygous carrier donors: Pediatr Transplant, 2017; 21(1) petr.12848
13. Wakiya T, Sanada Y, Urahashi T, Living donor liver transplantation from an asymptomatic mother who was a carrier for ornithine transcarbamylase deficiency: Pediatr Transplant, 2012; 16(6); E196-200
14. Sood V, Squires JE, Mazariegos GV, Living related liver transplantation for metabolic liver diseases in children: J Pediatr Gastroenterol Nutr, 2021; 72(1); 11-17
15. Oishi K, Arnon R, Wasserstein MP, Liver transplantation for pediatric inherited metabolic disorders: Considerations for indications, complications, and perioperative management: Pediatr Transplant, 2016; 20(6); 756-69
16. Darwish AA, McKiernan P, Chardot C, Paediatric liver transplantation for metabolic disorders. Part 1: Liver-based metabolic disorders without liver lesions: Clin Res Hepatol Gastroenterol, 2011; 35(3); 194-203
17. Darwish AA, McKiernan P, Chardot C, Paediatric liver transplantation for metabolic disorders. Part 2: Metabolic disorders with liver lesions: Clin Res Hepatol Gastroenterol, 2011; 35(4); 271-80
18. Moini M, Mistry P, Schilsky ML, Liver transplantation for inherited metabolic disorders of the liver: Curr Opin Organ Transplant, 2010; 15(3); 269-76
19. Yamada N, Sanada Y, Hirata Y, Selection of living donor liver grafts for patients weighing 6kg or less: Liver Transpl, 2015; 21(2); 233-38
20. Mizuta K, Yasuda Y, Egami S, Living donor liver transplantation for neonates using segment 2 monosubsegment graft: Am J Transplant, 2010; 10(11); 2547-52
21. Yamada N, Inui A, Sanada Y, Pediatric liver transplantation for neonatal-onset Niemann-Pick disease type C: Japanese multicenter experience: Pediatr Transplant, 2019; 23(5); e13462
22. Okada N, Ihara Y, Urahashi T, Antenatal immunoglobulin for prevention of neonatal hemochromatosis: Pediatr Int, 2016; 58(10); 1059-61
23. Kasahara M, Horikawa R, Tagawa M, Current role of liver transplantation for methylmalonic acidemia: A review of the literature: Pediatr Transplant, 2006; 10(8); 943-47
24. Morioka D, Ksahara M, Horikawa R, Efficacy of living donor liver transplantation for patients with methylmalonic acidemia: Am J Transplant, 2007; 7(12); 2782-87
25. Spada M, Calvo PL, Brunati A, Early liver transplantation for neonatal-onset methylmalonic acidemia: Pediatrics, 2015; 136(1); e252-56
26. Mallea J, Bolan C, Cortese C, Cystic fibrosis-associated liver disease in lung transplant recipients: Liver Transpl, 2019; 25(8); 1265-75
27. Plöchl W, Spiss CK, Plöchl E, Death after transplantation of a liver from a donor with unrecognized ornithine transcarbamylase deficiency: N Engl J Med, 1999; 341(12); 921-22
28. Ramanathan M, Uppalapu S, Patel NM, Hiding in plain sight: A case of ornithine transcarbamylase deficiency unmasked post-liver transplantation: Am J Transplant, 2017; 17(5); 1405-8
29. Lee CH, Ellaway C, Shun A, Split-graft liver transplantation from an adult donor with an unrecognized UCD to a pediatric and adult recipient: Pediatr Transplant, 2018; 22(1) petr.13073
30. Okada N, Sasaki A, Saito J, The Japanese experience and pharmacokinetics of antenatal maternal high-dose immunoglobulin treatment as a prophylaxis for neonatal hemochromatosis in siblings: J Matern Fetal Neonatal Med, 2020; 33(1); 142-48
31. McKiernan PJ, Squires JE, Squires RH, Liver transplant for inherited metabolic disease among siblings: Clin Transplant, 2020; 34(11); e14090
32. Kido J, Matsumoto S, Häberle J, Role of liver transplantation in urea cycle disorders: Report from a nationwide study in Japan: J Inherit Metab Dis, 2021 [Online ahead of print]
33. Kido J, Matsumoto S, Momosaki K, Liver transplantation may prevent neurodevelopmental deterioration in high-risk patients with urea cycle disorders: Pediatr Transplant, 2017; 21(6) petr.12987
34. Kido J, Matsumoto S, Mitsubuchi H, Early liver transplantation in neonatal-onset and moderate urea cycle disorders may lead to normal neurodevelopment: Metab Brain Dis, 2018; 33(5); 1517-23
Tables
Table 1. Demographic data for recipients and graft information.Table 2. Univariate analysis of risk factors for post-transplant complications in recipients with IMDs and BA.Table 3. Outcomes of LDLT using grafts from OTCD heterozygous carrier donors.Table 4. Outcomes of LDLT for patients with MMA in our institution.Table 5. Post-transplant survival rates of patients with IMDs.Table 1. Demographic data for recipients and graft information.Table 2. Univariate analysis of risk factors for post-transplant complications in recipients with IMDs and BA.Table 3. Outcomes of LDLT using grafts from OTCD heterozygous carrier donors.Table 4. Outcomes of LDLT for patients with MMA in our institution.Table 5. Post-transplant survival rates of patients with IMDs. In Press
15 Mar 2024 : Review article
Approaches and Challenges in the Current Management of Cytomegalovirus in Transplant Recipients: Highlighti...Ann Transplant In Press; DOI: 10.12659/AOT.941185
18 Mar 2024 : Original article
Does Antibiotic Use Increase the Risk of Post-Transplantation Diabetes Mellitus? A Retrospective Study of R...Ann Transplant In Press; DOI: 10.12659/AOT.943282
20 Mar 2024 : Original article
Transplant Nephrectomy: A Comparative Study of Timing and Techniques in a Single InstitutionAnn Transplant In Press; DOI: 10.12659/AOT.942252
28 Mar 2024 : Original article
Association Between FEV₁ Decline Rate and Mortality in Long-Term Follow-Up of a 21-Patient Pilot Clinical T...Ann Transplant In Press; DOI: 10.12659/AOT.942823
Most Viewed Current Articles
05 Apr 2022 : Original article
Impact of Statins on Hepatocellular Carcinoma Recurrence After Living-Donor Liver TransplantationDOI :10.12659/AOT.935604
Ann Transplant 2022; 27:e935604
12 Jan 2022 : Original article
Risk Factors for Developing BK Virus-Associated Nephropathy: A Single-Center Retrospective Cohort Study of ...DOI :10.12659/AOT.934738
Ann Transplant 2022; 27:e934738
22 Nov 2022 : Original article
Long-Term Effects of Everolimus-Facilitated Tacrolimus Reduction in Living-Donor Liver Transplant Recipient...DOI :10.12659/AOT.937988
Ann Transplant 2022; 27:e937988
15 Mar 2022 : Case report
Combined Liver, Pancreas-Duodenum, and Kidney Transplantation for Patients with Hepatitis B Cirrhosis, Urem...DOI :10.12659/AOT.935860
Ann Transplant 2022; 27:e935860