05 October 2021: Original Paper
Wilson Disease in Children; Chelation Therapy or Liver Transplantation? A 10-Year Experience from Pakistan
Sahira Aaraj1CDE*, Sabeen Abid KhanDOI: 10.12659/AOT.932606
Ann Transplant 2021; 26:e932606






Abstract
BACKGROUND: Wilson disease (WD) is a rare genetic disorder with vast clinical presentations and a higher incidence in areas where consanguinity is common. Most patients can be treated with oral chelation, but some require advanced surgical intervention, like liver transplantation (LT). This study aims to review outcomes of WD patients presenting to a tertiary care center over a period of 10 years.
MATERIAL AND METHODS: This retrospective analysis was conducted at Shifa International Hospital, Islamabad, Pakistan. Patients <18 years who were diagnosed with WD per ESPAGHAN guidelines from 2010 to 2020 were included. Presentation, diagnosis, treatment, and LT and its complications were recorded. Follow-ups were recorded, and patients were contacted by phone in cases of interrupted follow-up. Frequencies and percentages of variables were calculated.
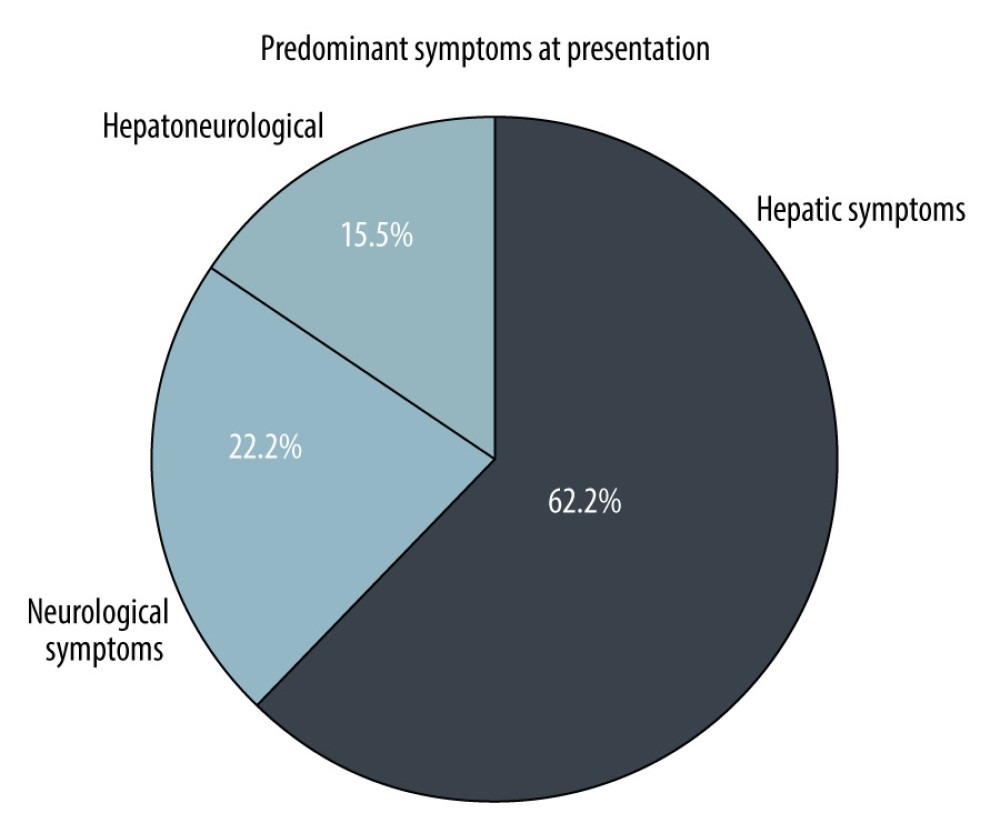
RESULTS: A total of 48 patients with WD were identified. Symptomatic disease was seen in 45 patients, with 3 diagnosed on screening. The hepatic form was common (62.2%). Mean age at diagnosis was 9.74 (range 5-17) years, 28 (58.3%) were male, while 17 (35.4%) were female. Urinary copper was increased in all patients (645.82±528.40). Oral treatment with penicillamine was given to 34 (75.5%) patients; 4 (8.9%) died while on oral treatment. Living donor LT was performed in 11 (22.9%) patients, who had a mean King’s Wilson index of 11 (range, 6-14). Currently, all LT patients are alive, with maximum graft survival of 7 years.
CONCLUSIONS: LT offers a promising treatment with good outcomes in pediatric WD. However, timely diagnosis and management with oral chelation therapy can prolong survival without LT.
Keywords: ((2,3,7,8,12,13,17,18)-octaethylporphinato)copper (II), Acute-On-Chronic Liver Failure, Hepatolenticular Degeneration, Hospitals, Pediatric, Liver Transplantation, Adolescent, Chelation Therapy, Child, Child, Preschool, Female, Humans, Pakistan
Background
Wilson disease (WD) is an autosomal recessive disease of copper metabolism, which was described by Wilson in 1912 [1]. The global prevalence of WD is 1 in 10 000 to 1 in 30 000 [1,2].
In Pakistan and neighboring countries like India and Bangladesh, there are no community-based prevalence studies of WD to date. The incidence of WD may be higher in Asia than in other world regions. Among pediatric liver diseases, WD accounts for 7.6% to 19.7% of cases in tertiary hepatobiliary centers, and there are 15 to 20 new cases of WD registered annually in the referral neurology centers of India [1].
The clinical manifestations of WD are diverse. In the first decade of life, WD presents with hepatic manifestations. After the age of 20 years, 75% of patients present with neurological manifestations and 25% with both hepatic and neuropsychiatric manifestations [1,2].
Owing to the relative rarity of WD and lack of awareness among physicians, the disease is not diagnosed early in most cases. Therefore, many patients present late with either advanced liver disease or incapacitating neurologic disease [3].
The European Society for Paediatric Gastroenterology, Hepatology and Nutrition guidelines advocate use of the Leipzig Scoring system for the diagnosis of WD. This includes the combination of clinical, biochemical, and genetic testing. Individual components include the presence or absence of Kayser-Fleischer (KF) rings, neurological symptoms, level of serum ceruloplasmin, presence of Coombs-negative hemolytic anemia, liver copper quantification, urinary copper excretion over 24 h, and mutation analysis for ATP7B mutations [4,5].
Medical management includes D-penicillamine, trientine, and zinc salts [6]. The aim of treatment is normalization of liver functions. The target urine copper excretion with a copper chelator is 200 to 500 mg/24 h. While on zinc monotherapy, the dosage should be 30 to 75 μg/day [3,5,6]. The normalization of non-ceruloplasmin-bound copper can be a useful parameter to monitor treatment response [7]. Dietary restriction of copper-rich foods is advised until remission of symptoms and normalization of liver enzymes in children treated with copper-chelating agents [6].
Research on new treatment modalities and gene/cell therapy in WD is ongoing. However, tetrathiomolybdate (bis-choline salt) is the only treatment in phase III development. Methanobactin and curcumin look promising as new and potentially safe treatment options; however, they have not yet been clinically tested [8,9].
Regarding medical management, disease prognosis is very good, with 85% survival over 5 decades, as reported by Schilsky et al [10]. However, in patients with liver failure and in selected patients with neurologic WD that is resistant to anti-copper therapies, liver transplantation (LT) is a lifesaving procedure [3].
The King’s Wilson index should be monitored for the prognostic assessment and timely decision-making for LT [4,6,7]. Chelating agents or zinc treatment are no longer required after transplantation.
Although LT for WD is a potentially lifesaving treatment, it is not free from complications. Shifa International Hospital, Islamabad is among the first institutes in Pakistan offering pediatric LT and gastroenterology services for various liver disorders. This study aims to evaluate the outcomes of pediatric WD, including the study of living donor LT for WD.
Material and Methods
This retrospective analysis was conducted at Shifa International Hospital from 2010 to 2020. After Investigational Review Board approval from Shifa International Hospital and Shifa Tameer-e-Millat University (no. 187-1007-2020), all patients under 18 years of age who were diagnosed with WD were included in the study. Diagnosis of WD was made according to the scoring system of Ferenci et al [11]. The diagnostic inclusion criteria of WD were increased (>40 μg) 24-h urinary copper, low serum ceruloplasmin (<20 mg/dL), KF rings, and evidence of Coombs-negative hemolytic anemia. A score of ≥4 was considered diagnostic.
Patient information was recorded on a self-designed proforma, including sex, age at presentation, anthropometry, and predominant system involved (hepatic or neurological). Hepatic symptoms included acute or chronic liver disease. Neurological WD included dystonia dysarthria, seizures, deterioration of school performance, and various personality and behavior changes. Various laboratory values were collected: hemoglobin, white cell count, serum ceruloplasmin level, total bilirubin, alanine aminotransferase, aspartate transaminase, prothrombin time/international normalized ratio values, serum albumin, creatinine, and 24-h urinary copper level. Information regarding oral treatment, diet, and water sources was recorded. Data on the preferred chelating agent used and adverse effects were collected. The King’s Wilson index was calculated.
The post-transplantation profile was reviewed for the indication of LT (acute, chronic, or acute-on-chronic disease), donor relation, length of Intensive Care Unit (ICU) stay, rejection (yes/no), duration of graft survival, drugs for immunosuppression, complications, and patient survival, death, or loss to follow-up.
The data were analyzed using SPSS version 23.0. Frequencies and percentages were calculated for the variables. Survival time was calculated by subtracting the date of death or last follow-up from the date of LT.
Results
OUTCOMES OF LT:
A total of 12 patients were advised to have LT, but 11 patients (22.9%) underwent the surgery. All cases were of living donor LT. All patients had hepatic symptoms. Seven (63.63%) patients had acute liver failure at presentation, while 4 (36.36%) presented with decompensated chronic liver disease. Six patients had a King’s Wilson index of more than 11 (range, 6–14). Patients with an index of <11 had presented with acute liver failure and end-stage liver disease. Table 3 shows the King’s Wilson indices of all patients.
Other laboratory values are shown in Table 4. Seven patients had an ICU stay of 1 week or less, while 3 were in the ICU for longer than 1 week.
Tacrolimus, used in 6 (54.5%) patients, was the most common drug used for immunosuppression, while cyclosporine was used in 3 (27.3%) patients. Tacrolimus was also used in combination with mycophenolate mofetil in 2 patients. To date, none of the patients have had recurrence of hepatic symptoms or graft rejection. No patients were administered penicillamine or any other chelation therapy after transplantation.
Immediate complications after transplantation were chest infection in 2 patients, biliary strictures in 3 patients, which required dilatation, and thrombosis in 1 patient. One patient developed ataxia, which resolved on medication. One of the patients developed a Candida infection. On long-term follow-up, 1 patient developed tuberculosis 1 year after transplantation, which was treated for 1 year with antitubercular treatment. The same patient developed biliary stricture 4 years after transplantation, for which percutaneous transhepatic biliary drainage was done. To date, all transplant patients are alive and doing well, with a maximum graft survival of 7 years.
Discussion
This study describes the outcomes of pediatric patients with WD receiving oral chelation or LT over a period of 10 years in a low-resource setting. As the first pediatric liver transplant center in Pakistan, we receive referrals from all over the country. The mean age of our patients was 9.7 years. Hepatic presentation (62.2%) of WD was more common than neurological WD (22.2%). Studies also support the fact that up to half of patients with WD present with hepatic disease [12].
The diagnostic workups for WD showed low serum ceruloplasmin in 69.8% of patients. Serum ceruloplasmin of less than 20 mg/dL has good accuracy in diagnosing WD [13]. According to previous reports, a low ceruloplasmin level has a sensitivity of 77% to 99% and specificity of 55% to 88.2% [14]. In the present study, 24-h urinary copper levels were elevated in all patients (100%), with a mean value of 645 mg/dL [15]. Together, high urinary copper and low serum ceruloplasmin have good diagnostic accuracy in diagnosing WD [16].
KF rings were seen in 14 (50%) patients with hepatic and 8 (80%) patients with neurological disease. Nagral et al also documented that KF rings are seen in 50% to 60% of hepatic and 95% to 100% of neurological WD cases [1]. In the present study, genetic testing was not done owing to the lack of availability in Pakistan. Genetic testing can help in mutation analysis and to identify asymptomatic patients and carriers [17]. The Leipzig scoring system could not be strictly followed in the present study owing to the lack of availability of liver biopsy and genetic/mutation analysis in most of our patients.
Our study confirmed the efficacy of chelation therapy in WD. The management protocol was uniform in all 34 patients undergoing chelation. Penicillamine with zinc therapy was the treatment choice in all cases. The efficacy of penicillamine varies from 50% to 66.6% [18]. In mild disease, its efficacy is 79% to 90% [3]. Of the 34 symptomatic children, 30 are alive to date, with a maximum age of 26 years. Only 2 (5.88%) of them were started on trientine because of bone marrow suppression. To date, over a mean period of 10 years, none of the patients in the present study who underwent chelation therapy have required LT. Our results are supported by studies from neighboring countries like India, where chelation therapy with penicillamine has shown good results, even in patients with severe hepatic disease [19]. Another study from India reported the adjunctive role of zinc therapy in pediatric WD, with good outcomes [20].
We had 11 patients with WD who underwent LT. Fulminant hepatic failure was seen in 7 (63.6%) patients, while 4 (36.3%) patients had decompensated chronic liver disease. Lankarani et al, from Iran, described a much larger cohort of 107 patients with WD who underwent LT; 21 (19.6%) patients had fulminant hepatic failure, while 86 (80.3%) had chronic WD [21]. The literature supports that LT remains the definite treatment modality for the complete correction of liver defects in WD [22]. LT also offers good short-term and long-term survival, with limited complications [23]. Studies have also highlighted better survival in children than in adults receiving living donor LT [24], as was seen in our cohort of patients.
Post-transplant complications experienced by our patients included infection (2 patients), biliary stricture (3 patients), and thrombosis (1 patient). Elisofon et al described the first 30 days of surgical complications, including reoperation in 31.7% of patients, and biliary tract complications, including strictures in 13.6%, hepatic artery thrombosis in 6.35%, and portal vein thrombosis in 3.2% of patients [25].
We did not have any patients with neurological WD who underwent LT; however, Poujois et al reported 18 patients with neurological WD who underwent LT, reporting a 5-year survival of 72.2%, with major improvement in symptoms [26].
In the present study, we have had 100% survival at 7 years after transplantation. This is encouraging, as the literature from developed countries report a 5-year survival rate of up to 85% [27]. Table 5 compares the outcomes of LT for WD.
Conclusions
Liver transplantation offers a promising treatment with very good outcomes in pediatric patients with acute liver failure or decompensated liver disease secondary to WD [26]. However, timely diagnosis and management with oral chelation therapy can prolong survival without LT. The affordability and access to LT remains an issue in developing countries like Pakistan.
Tables
Table 1. Summary of symptoms of patients with Wilson disease.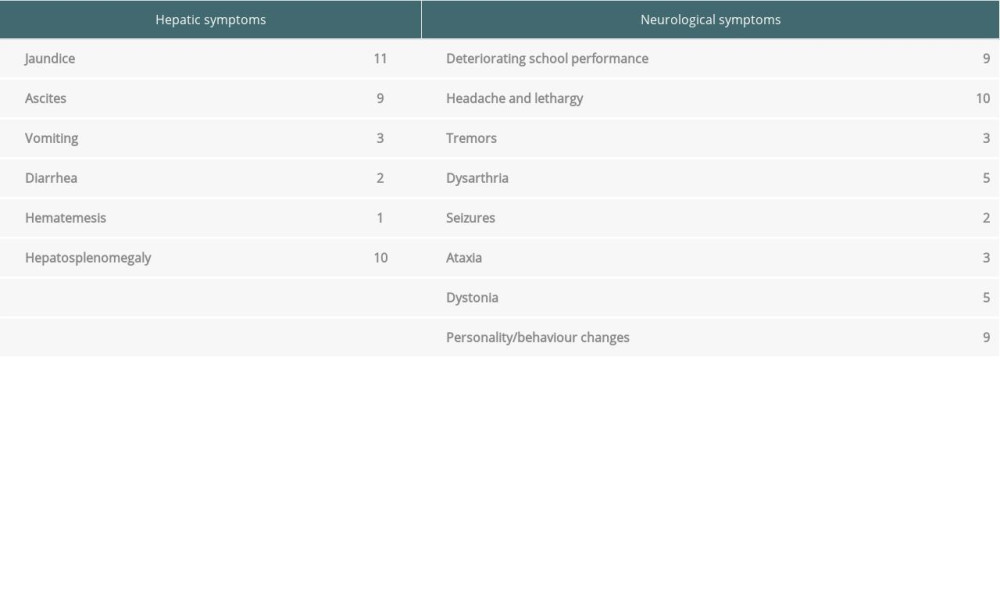 Table 2. Demographic, clinical, and laboratory data of patients with Wilson disease.
Table 2. Demographic, clinical, and laboratory data of patients with Wilson disease.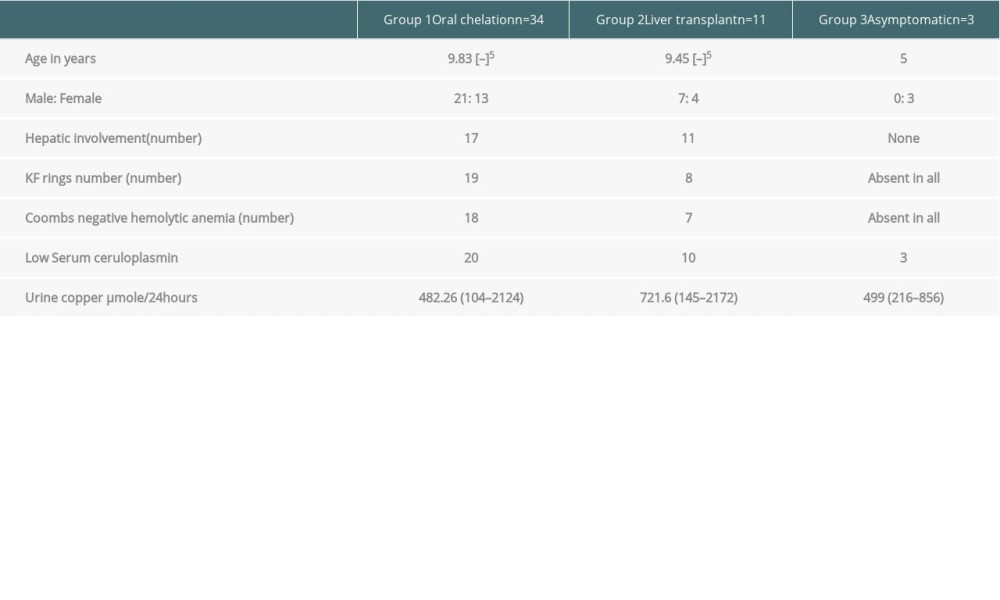 Table 3. Laboratory indices of patients with Wilson disease (mean values).
Table 3. Laboratory indices of patients with Wilson disease (mean values).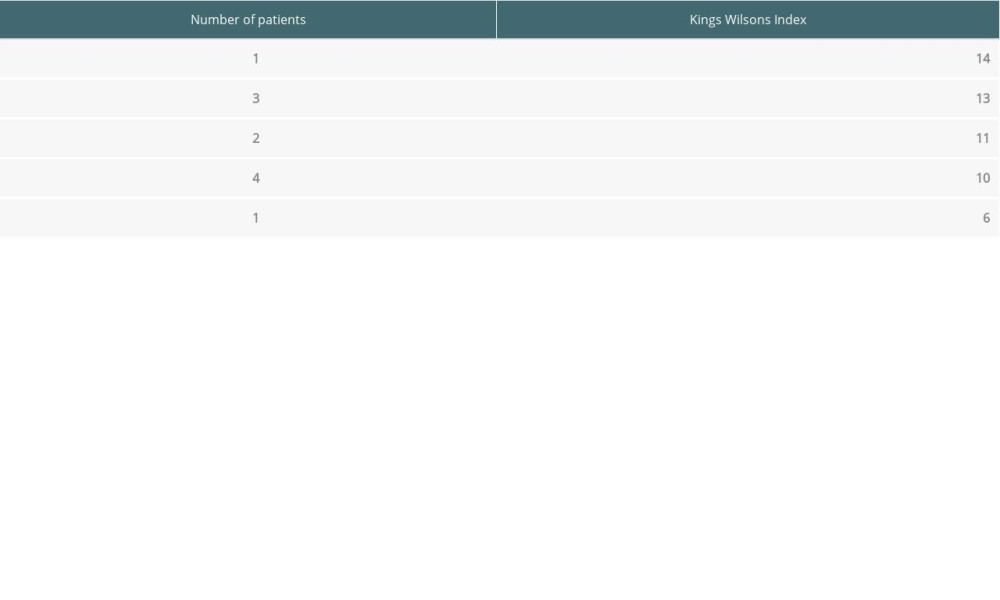 Table 4. King’s Wilson indices of individual patients with Wilson disease.
Table 4. King’s Wilson indices of individual patients with Wilson disease.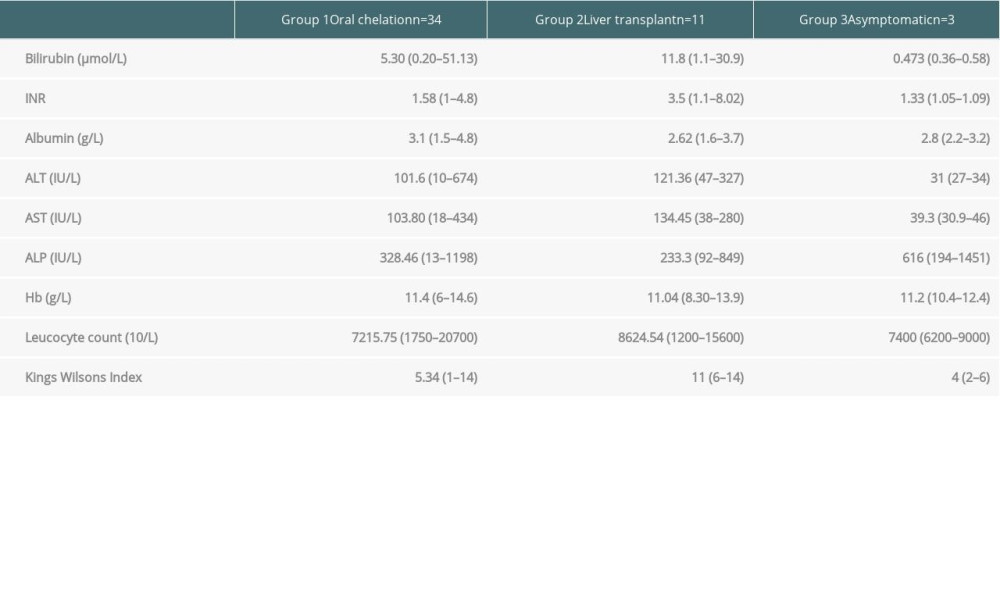 Table 5. Outcome of liver transplantation in patients with Wilson disease.
Table 5. Outcome of liver transplantation in patients with Wilson disease.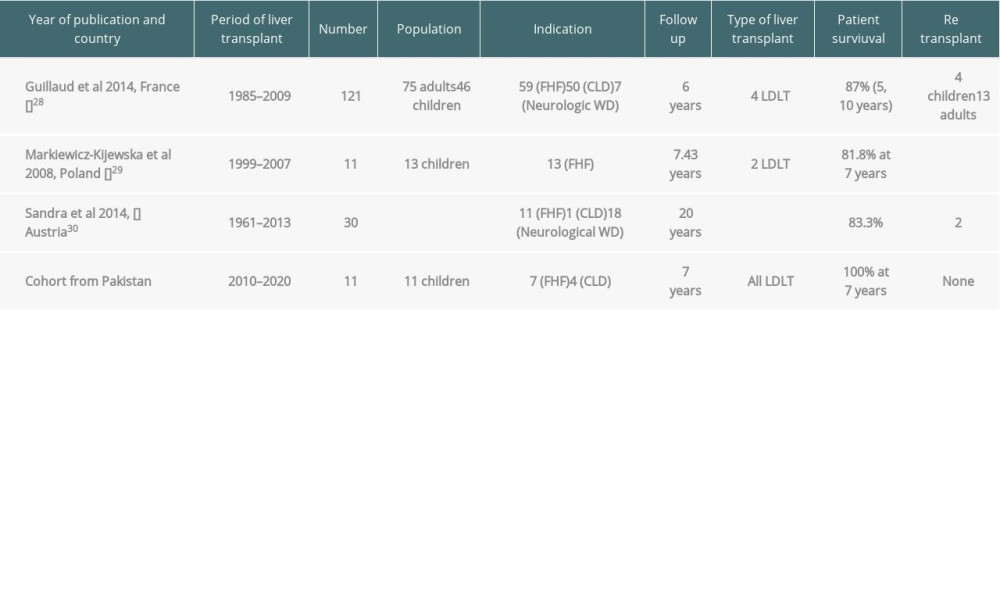
References
1. Nagral A, Sarma MS, Matthai J, Wilson’s disease: Clinical practice guidelines of the indian national association for study of the liver, the Indian Society of Pediatric Gastroenterology, Hepatology and Nutrition, and the Movement Disorders Society of India: J Clin Exp Hepatol, 2018; 9(1); 74-98
2. Hamdani SSB, Cheema HA, Saeed A, Electrocardiographic manifestations in pediatric Wilson disease: J Ayub Med Coll, 2018; 30(1); 22-25
3. Parkash O, Ayub A, Jafri W, Wilson’s disease: Experience at a tertiary care hospital: J Coll Physicians Surg Pak, 2013; 23(7); 525-26
4. Palumbo CS, Schilsky ML, Clinical practice guidelines in Wilson disease: Ann Transl Med, 2019; 7(Suppl 2); S65
5. Pfister ED, Karch A, Adam R, Predictive factors for survival in children receiving liver transplant for Wilsons disease: A cohort study using European liver transplant registry data: Liver Transplant, 2018; 24; 1186-98
6. Socha P, Janczyk W, Dhawan A, Wilsons disease in children: A position paper by the Hepatology committee of the European Society for pediatric gastroenterology, Hepatology and Nutrition: J Pediatr Gastroenterol Nutr, 2018; 66; 334-44
7. , EASL Clinical Practice Guidelines: Wilson’s disease. European Association for the Study of the Liver: J Hepatol, 2012; 56; 671-85
8. Kathawala M, Hirschfield GM, Insights into the management of Wilson’s disease: Therap Adv Gastroenterol, 2017; 10(11); 889-905
9. Litwin T, Dzieżyc K, Członkowska A, Wilson disease – treatment perspectives: Ann Transl Med, 2019; 7(Suppl 2); S68
10. Schilsky ML, Long-term outcome for Wilson disease: 85% good: Clin Gastroenterol Hepatol, 2014; 12; 690-99
11. Ferenci P, Caca K, Loudianos G, Diagnosis and phenotypic classification of Wilson disease: Liver Int, 2003; 23; 139-42
12. Guindi M, Wilson disease: Semin Diagn Pathol, 2019; 36(6); 415-22
13. Kim JA, Kim HJ, Cho JM, Diagnostic value of ceruloplasmin in the diagnosis of pediatric Wilson’s Disease: J Pediatr Gastroenterol Nutr, 2015; 18(3); 187-92
14. Ryan A, Nevitt SJ, Tuohy O, Cook P, Biomarkers for diagnosis of Wilson’s disease: Cochrane Database Syst Rev, 2019; 2019(11); CD012267
15. Vieira J, Oliveira PV, Juliano Y, Urinary copper excretion before and after oral intake of d-penicillamine in parents of patients with Wilson’s disease: Dig Liver Dis, 2012; 44(4); 323-27
16. Aksu AÜ, Sarı S, Gürkan ÖE, Dalgıç B, Urinary 24-hour copper excretion at the time of diagnosis in children with Wilson’s disease: Acta Gastroenterol Belg, 2018; 81(3); 410-14
17. Li X, Lu Z, Lin Y, Clinical features and mutational analysis in 114 young children with Wilson disease from South China: Am J Med Genet A, 2019; 179(8); 1451-58
18. Rukunuzzaman M, Karim AB, Nurullah M, Childhood Wilson disease: Bangladesh perspective: Mymensingh Med J, 2017; 26(2); 406-13
19. Das MC, Sarma MS, Srivastav A, Effect of chelation therapy in pediatric Wilson’s disease: Liver and endoscopic outcome: J Hepatobiliary Pancreat Sci, 2021; 28; 336-45
20. Gupta P, Choksi M, Goel A, Maintenance zinc therapy after initial penicillamine chelation to treat symptomatic hepatic Wilsons disease in resource constrained setting: Indian J Gastroenterol, 2018; 37(1); 310-38
21. Lankarani KB, Malek-Hosseini SA, Nikeghbalian S, Fourteen years of experience of liver transplantation for Wilson’s Disease; A report on 107 cases from Shiraz, Iran: PLoS One, 2016; 11(12); e0167890
22. Sood V, Squires JE, Mazariegos GV, Living related liver transplantation for metabolic liver diseases in children: J Pediatr Gastroenterol Nutr, 2021; 72(1); 11-17
23. Pham YH, Miloh T, Liver transplantation in children: Clin Liver Dis, 2018; 22(4); 807-21
24. Mogul DB, Lee J, Purnell TS, Barriers to access in pediatric living-donor liver transplantation: Pediatr Transplant, 2019; 23(6); e13513
25. Elisofon SA, Magee JC, Ng VL, Society of pediatric liver transplantation: Current registry status 2011–2018: Pediatr Transplant, 2020; 24(1); e13605
26. Poujois A, Sobesky R, Meissner WG, Liver transplantation as a rescue therapy for severe neurologic forms of Wilson disease: Neurology, 2020; 94(21); e2189-202
27. Cortes M, Heaton ND, Dhawan A, Liver transplantation in children: State of the art and future perspectives: Arch Dis Child, 2017; 103(2); 192-98
28. Guillaud O, Dumortier J, Sobesky R, Long term results of liver transplantation for Wilson’s disease: experience in France: J Hepatol, 2014; 60(3); 579-89
29. Markiewicz-Kijewska M, Szymczak M, Ismail H, Liver transplantation for fulminant Wilson’s disease in children: Ann Transplant, 2008; 13(2); 28-31
30. Beinhardt S, Leiss W, Stattermayer AF, Long-term outcomes of patients with Wilson disease in a large Austrian cohort: Clin Gastroenterol Hepatol, 2014; 12(4); 683-89
Tables
Table 1. Summary of symptoms of patients with Wilson disease.Table 2. Demographic, clinical, and laboratory data of patients with Wilson disease.Table 3. Laboratory indices of patients with Wilson disease (mean values).Table 4. King’s Wilson indices of individual patients with Wilson disease.Table 5. Outcome of liver transplantation in patients with Wilson disease.Table 1. Summary of symptoms of patients with Wilson disease.Table 2. Demographic, clinical, and laboratory data of patients with Wilson disease.Table 3. Laboratory indices of patients with Wilson disease (mean values).Table 4. King’s Wilson indices of individual patients with Wilson disease.Table 5. Outcome of liver transplantation in patients with Wilson disease. In Press
15 Mar 2024 : Review article
Approaches and Challenges in the Current Management of Cytomegalovirus in Transplant Recipients: Highlighti...Ann Transplant In Press; DOI: 10.12659/AOT.941185
18 Mar 2024 : Original article
Does Antibiotic Use Increase the Risk of Post-Transplantation Diabetes Mellitus? A Retrospective Study of R...Ann Transplant In Press; DOI: 10.12659/AOT.943282
20 Mar 2024 : Original article
Transplant Nephrectomy: A Comparative Study of Timing and Techniques in a Single InstitutionAnn Transplant In Press; DOI: 10.12659/AOT.942252
28 Mar 2024 : Original article
Association Between FEV₁ Decline Rate and Mortality in Long-Term Follow-Up of a 21-Patient Pilot Clinical T...Ann Transplant In Press; DOI: 10.12659/AOT.942823
Most Viewed Current Articles
05 Apr 2022 : Original article
Impact of Statins on Hepatocellular Carcinoma Recurrence After Living-Donor Liver TransplantationDOI :10.12659/AOT.935604
Ann Transplant 2022; 27:e935604
12 Jan 2022 : Original article
Risk Factors for Developing BK Virus-Associated Nephropathy: A Single-Center Retrospective Cohort Study of ...DOI :10.12659/AOT.934738
Ann Transplant 2022; 27:e934738
22 Nov 2022 : Original article
Long-Term Effects of Everolimus-Facilitated Tacrolimus Reduction in Living-Donor Liver Transplant Recipient...DOI :10.12659/AOT.937988
Ann Transplant 2022; 27:e937988
15 Mar 2022 : Case report
Combined Liver, Pancreas-Duodenum, and Kidney Transplantation for Patients with Hepatitis B Cirrhosis, Urem...DOI :10.12659/AOT.935860
Ann Transplant 2022; 27:e935860